
VYVGART® (Efgartigimod alfa) ist seit Juni 2025 in Deutschland für die Chronisch inflammatorische demyelinisierende Polyradikuloneuropathie (CIDP) zugelassen und wird als Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver CIDP nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet.
Bereits seit 2022 steht es für die Therapie der generalisierte Myasthenia gravis (gMG) für Patient:innen, die Anti-Acetylcholin-Re zeptor (AChR)-Antikörper positiv sind, zur Verfügung. Das IgG1-Antikörper-Fc-Fragment VYVGART® (Efgartigimod alfa) wurde speziell für die Reduktion pathogener IgG-Autoantikörper entwickelt.1,2
VYVGART® – Fortschritt in der CIDP-Therapie nach über 30 Jahren
Mit VYVGART® steht erstmals seit drei Jahrzehnten ein neuer Wirkmechanismus zur Behandlung der CIDP zur Verfügung, der gezielt und passgenau an FcRn bindet. Die Zulassungsstudie ADHERE, die bislang größte klinische Studie in diesem Krankheitsbild, belegt als primären Endpunkt in der Phase A eine hohe Ansprechrate sowie eine signifikante Reduktion des Rückfallrisikos in Phase B. Darüber hinaus zeigte sich VYVGART® als gut verträglich. Die einfache Anwendung als Fertigspritze macht VYVGART® besonders alltagsfreundlich und unterstützt Patient:innen dabei, ihre Therapie einfach und flexibel in den Alltag zu integrieren.3
Damit setzt VYVGART® neue Maßstäbe in der Versorgung von CIDP-Betroffenen.
Screening und Run-in-Phase3
SCREENING: ≤ 4 WOCHEN
Bestätigung der CIDP-Diagnose
RUN-IN: ≤ 12 WOCHEN
Bestätigung einer aktiven Erkrankung
Patient:innen mit bestätigter aktiver Erkrankung
Phase A: Initiale Behandlung
Open label - Dauer ≤ 12 Wochen
VYVGART® (n = 322)
Wöchentliche Gabe 1.000 mg s.c.
Responder3
Patient:innen mit nachgewiesener klinischer Verbesserung bei zwei aufeinanderfolgenden Untersuchungen**
Responder
Phase B: Randomisierte Behandlung
Doppelblind, placebokontrolliert – Dauer ≤ 48 Wochen
VYVGART®
Wöchentliche Gabe 1.000 mg s.c.
vs.
Placebo (n = 110)
Wöchentliche Gabe s.c.*
Primärer Endpunkt3
Zeit bis zur ersten klinischen Verschlechterung im Vergleich zur Baseline der Phase B
Ergebnisse der Gesamtpopulation
Die hier gezeigten Daten zeigen die gesamte ADHERE-Studienpopulation. VYVGART® ist zugelassen für die Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen.
* 2.000 U/ml Hyaluronidase s.c. Q1W.
**definiert als Verbesserung (≥ 1 Punkt Abnahme) des aINCAT-Scores im Vergleich zum Ausgangswert der Phase A. Bei Teilnehmer:innen, die keine Veränderung des aINCAT-Scores aufwiesen und bei denen sich der I-RODS-Score und/oder die Griffstärke während der Run-in-Phase verschlechterte, wurde die klinische Verbesserung als I-RODS-Verbesserung von ≥ 4 Punkten und/oder ein Verbesserung der Griffstarke von ≥ 8 kPa während der Phase A oder als aINCAT-Verbesserung definiert.3
Die ADHERE-Studie begann mit einer Screening-Phase von bis zu 4 Wochen.
Im Rahmen des Screenings wurde geprüft, ob die Teilnehmenden eine dokumentierte, bestätigte oder vermutete Diagnose einer progressiven oder rezidivierenden CIDP nach den Kriterien der European Federation of Neurological Societies (EFNS) und der Peripheral Nerve Society (PNS) von 2010 aufwiesen.4
Die Diagnose galt als bestätigt, wenn zwei CIDP-Expert:innen unabhängig voneinander eine wahrscheinliche oder gesicherte CIDP gemäß den EFNS/PNS-Kriterien von 2010 feststellten. Sobald die Diagnose einer CIPD bestätigt war, konnten die Patient:innen mit der Run-in-Phase beginnen.
Nur Patient:innen mit bestätigter CIDP konnten in die Run-in-Phase der Studie aufgenommen werden.
Eingeschlossen wurden Patient:innen, die zum Zeitpunkt des Screenings in Behandlung waren und eine Standardtherapie erhielten (IVIg, SCIg oder Kortikosteroide) sowie Patient:innen, die für ≥ 6 Monate vor Studieneinschluss keine Standardbehandlung erhielten oder therapienaiv waren. Ausgeschlossen waren Patient:innen mit einer rein sensorischen CIDP-Variante.
Ziel der Run-in-Phase war, die Patient:innen zu identifizieren, die eine aktive CIDP aufwiesen. Aktive CIDP war dabei definiert als klinisch relevante Verschlechterung nach dem Absetzen der bisherigen Therapie, d.h. eine Zunahme um ≥ 1 Punkt im aINCAT, eine I-RODS-Abnahme um ≥ 4 Punkte (unter Verwendung der Zentil-Metrik) oder eine Abnahme der Handkraft um ≥ 8 kPa.
Zu Beginn der Run-in-Phase mussten die Patient:innen ihre Vortherapie daher abbrechen. Die Funktionsfähigkeit der Patient:innen wurde dann in den regelmäßigen Kontrollbesuchen, die für alle 4 Wochen geplant waren oder sobald Hinweise auf eine Verschlechterung vorlagen, bis zu 12 Wochen untersucht.
Patient:innen, die nach Absetzen der Behandlung die Kriterien für eine klinisch relevante Verschlechterung erfüllten, konnten die Run-in-Phase beenden und in Phase A wechseln.
Für den klinischen Alltag empfiehlt die Fachinformation VYVGART® 1000 mg Injektionslösung in einer Fertigspritze folgendes Vorgehen: Bei Patient:innen, die von ihren derzeitigen CIDP-Therapien umgestellt werden, sollte die Behandlung mit VYVGART® vorzugsweise eingeleitet werden, bevor die klinische Wirkung dieser vorherigen Therapien nachzulassen beginnt.2
X
Der adjustierte INCAT-Score (aINCAT) ist eine modifizierte Version den INCAT, die vor allem in klinischen Studien wie ADHERE verwendet wird. Im Gegensatz zum INCAT werden dabei nur nachhaltige, funktionell bedeutsam Veränderungen ≥ 1 Punkt gewertet, um einen Bias durch vorübergehende Fluktuationen zu vermeiden.
Phase A (open label) – Hohe Ansprechrate unter VYVGART®
Ziel der Phase A war es, sogenannte Responder zu erkennen, d.h. Patient:innen, die unter VYVGART® in zwei aufeinanderfolgenden Besuchen bei wöchentlicher Gabe eine klinische bestätigte funktionelle Verbesserung erreichten, und damit mindestens eines der folgenden Kriterien erfüllten:
1. Verbesserung des modifizierten INCAT-Scores um ≥ 1 Punkt
2. Anstieg des I-RODS-Zentilwerts um ≥ 4 Punkte
3. Zunahme der Handkraft (gemessen mit dem Martin-Vigorimeter) um ≥ 8 kPa
Die 322 Teilnehmenden mit aktiver CIDP erhielten dazu bis zu 12 Wochen lang 1000 mg VYVGART® s.c. wöchentlich.
In die offene Behandlungsphase wurden 322 Patient:innen mit aktiver CIDP eingeschlossen, darunter auch therapienaive sowie vorbehandelte Personen.
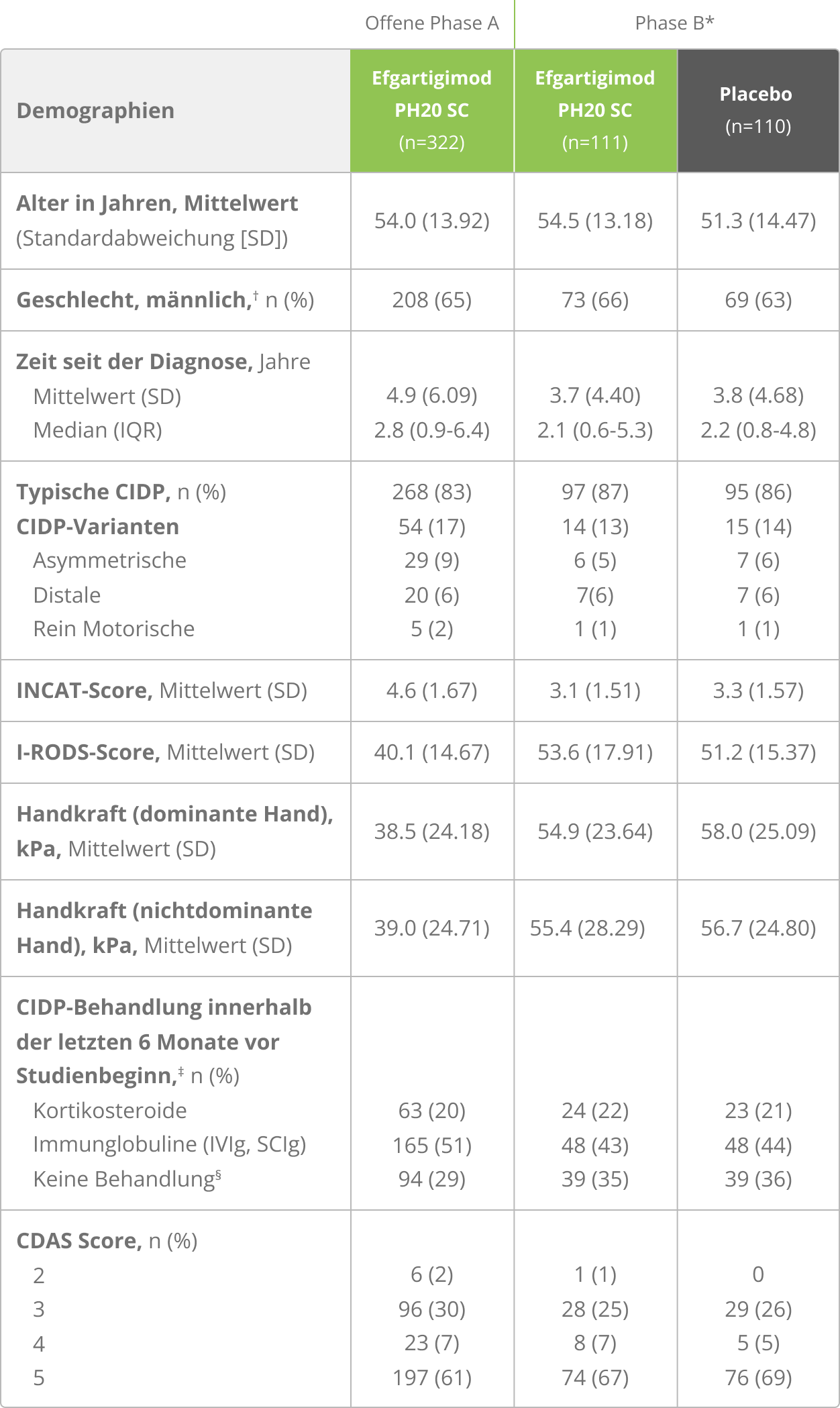
Ergebnisse der Gesamtpopulation
Die hier gezeigten Daten zeigen die gesamte ADHERE-Studienpopulation. VYVGART® ist zugelassen für die Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen.
* Klinische Bewertungen (INCAT, I-RODS und Handkraft) wurden zu Beginn jeder Phase durchgeführt, während die restlichen Ergebnisse im Rahmen des Screenings bewertet wurden.
Das Geschlecht der Teilnehmer:innen wurde als "Geschlecht bei der Geburt" (weiblich oder männlich) abgefragt und von dem/der Ärzt:in während des Screenings angegeben;
‡ Die Ausgangswerte der Phase B beruhen auf randomisierten Schichtungsfaktoren.
§ Keine Behandlung wurde definiert als Patient:innen, die noch nie eine CIDP-Behandlung erhalten hatten (Behandlungs-naiv) oder die innerhalb von 6 Monaten vor Studieneintritt keine CIDP-Behandlung (Kortikosteroide, IVIg oder SCIg) erhalten hatten.
VYVGART®: Hohe Ansprechrate – 66,5 % der Patient:innen erzielten eine klinische Verbesserung*,2,3
In der offenen Phase A der ADHERE-Studie erreichten 66,5 % der Patient:innen unter Efgartigimod alfa eine klinisch bestätigte funktionelle Verbesserung (primärer Endpunkt; 95 %-KI: 61,0–71,6 %).
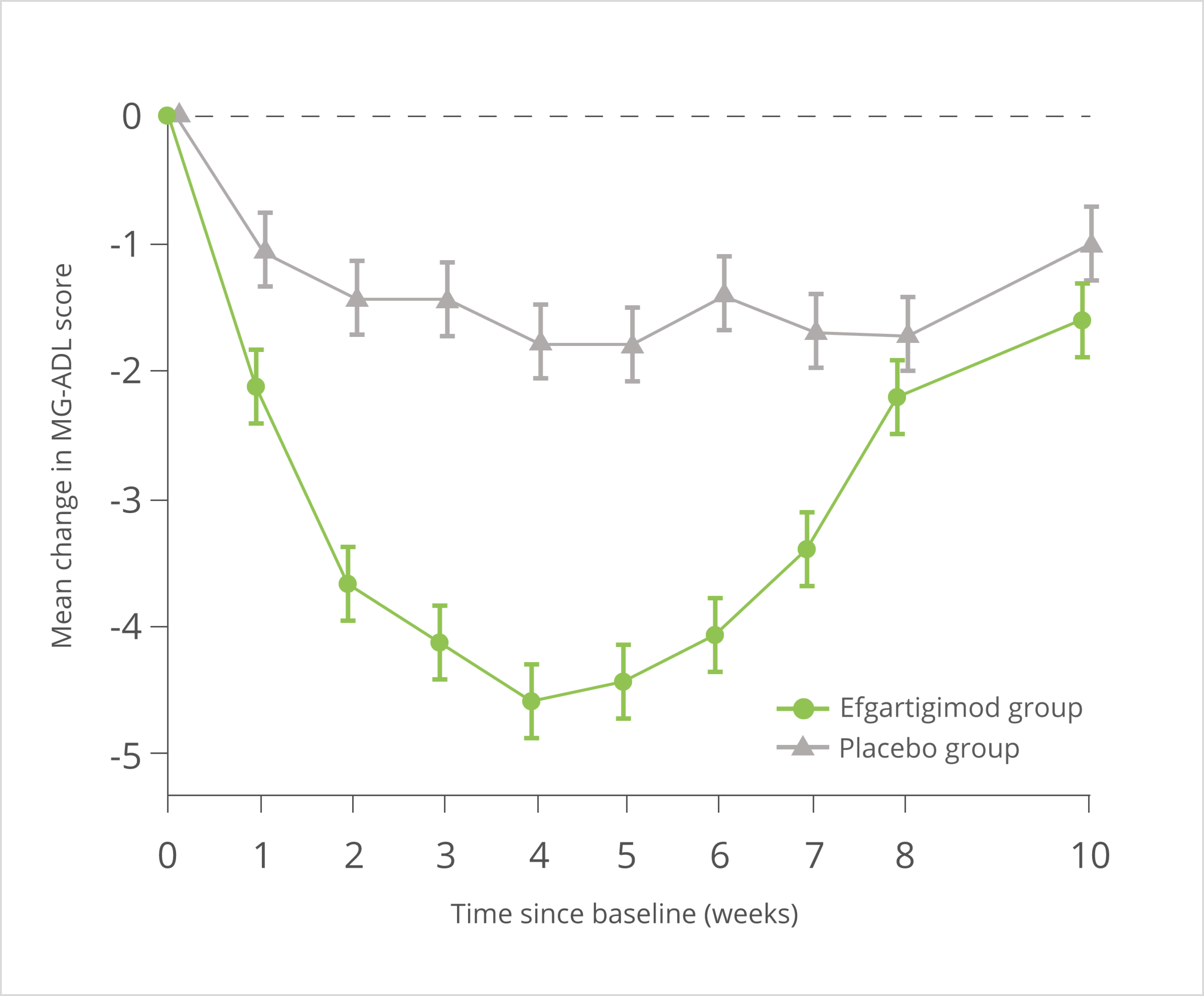
Ergebnisse der Gesamtpopulation
Die hier gezeigten Daten zeigen die gesamte ADHERE-Studienpopulation. VYVGART® wird als Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet.
* definiert als Verbesserung (≥ 1 Punkt Abnahme) des aINCAT-Scores im Vergleich zum Ausgangswert der Phase A. Bei Teilnehmer:innen, die keine Veränderung des aINCAT-Scores aufwiesen und bei denen sich der I-RODS-Score und/oder die Griffstärke während der Run-in-Phase verschlechterte, wurde die klinische Verbesserung als I-RODS-Verbesserung von ≥ 4 Punkten und/oder eine Verbesserung der Griffstärke von ≥ 8 kPa während der Phase A definiert.3
aINCAT: adjusted Inflammatory Neuropathy Cause and Treatment; I-RODS: Inflammatory Rasch-built Overall Disability Scale; KI: Konfidenzintervall; kPA: Kilopascal
In einer präspezifizierten Analyse der offenen Phase A der ADHERE-Studie wurde zudem untersucht, wann Patient:innen unter wöchentlicher Therapie mit VYVGART® erstmals eine klinische Verbesserung zeigten (sekundärer Endpunkt). Erste klinische Verbesserungen wurden bereits nach 9 Tagen bei 25 % der Patient:innen beobachtet. Innerhalb von 22 Tagen erreichten 50 % eine Besserung.3
Bei der Analyse des Sicherheitskollektivs, in der alle Patient:innen betrachtet wurden, die mindestens eine VYVGART®-Injektion erhalten hatten (Responder und Non-Responder) zeigte sich in der offenen Phase A der ADHERE-Studie im Mittel eine klinisch bedeutsame Verbesserung über alle untersuchten Endpunkte hinweg3:
- Funktionelle Verbesserung:
Der modifizierte INCAT-Score verbesserte sich im Mittel um 0,9 Punkte, bei Respondern sogar um 1,4 Punkte.
- Verbesserung der Lebensqualität:
Der I-RODS-Score stieg im Mittel um 7,7 Punkte, bei Respondern im Mittel um 12 Punkte, d.h. ein signifikanter Zugewinn an Selbstständigkeit im Alltag.
- Verbesserung der Handkraft:
Die Handkraft verbesserte sich um durchschnittlich 12,3 kPa (dominante Hand) bzw. 11,2 kPa (nichtdominante Hand), bei Respondern sogar um rund 18 kPa (dominante Hand) bzw. 17 kPa im Mittel (nichtdominante Hand).
- Motorische Leistung:
Der MRC-Gesamtscore nahm im Mittel um 3,8 Punkte (5,9 Punkte bei den Respondern) zu, die Mobilität (TUG-Test) verbesserte sich deutlich (-4,3 Sekunden bzw. -6,1 Sekunden bei Respondern).
Phase B (randomisierte Behandlung) – Signifikante Reduktion des relativen Rückfallrisikos unter VYVGART®
Alle Patient:innen mit einer klinischen bestätigten funktionellen Verbesserung unter VYVGART® aus Phase A (n=221) wurden anschließend in Phase B randomisiert mit VYVGART® (n=111) oder Placebo (n=110) über bis zu 48 Wochen weiterbehandelt. Der primäre Endpunkt der Phase B war die Zeit bis zur ersten klinischen Verschlechterung im aINCAT im Vergleich zur Baseline. Die Baseline-Charakteristika waren zwischen den beiden Armen ausgewogen.
Ergebnisse der Gesamtpopulation
Die hier gezeigten Daten zeigen die gesamte ADHERE-Studienpopulation. VYVGART® ist zugelassen für die Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen.
* Klinische Bewertungen (INCAT, I-RODS und Handkraft) wurden zu Beginn jeder Phase durchgeführt, während die restlichen Ergebnisse im Rahmen des Screenings bewertet wurden.
† Das Geschlecht der Teilnehmer:innen wurde als "Geschlecht bei der Geburt" (weiblich oder männlich) abgefragt und von dem/der Ärzt:in während des Screenings angegeben;
‡ Die Ausgangswerte der Phase B beruhen auf randomisierten Schichtungsfaktoren.
§ Keine Behandlung wurde definiert als Patient:innen, die noch nie eine CIDP-Behandlung erhalten hatten (Behandlungs-naiv) oder die innerhalb von 6 Monaten vor Studieneintritt keine CIDP-Behandlung (Kortikosteroide, IVIg oder SCIg) erhalten hatten.
Die randomisierte Phase B dauerte maximal 48 Wochen. Wenn Patient:innen eine klinische Verschlechterung (Rückfall) zeigten, endete ihre Teilnahme an der randomisierten Phase B.
Eine klinische Verschlechterung war definiert als ein Anstieg des aINCAT um ≥1 Punkt im Vergleich zu Baseline von Phase B, der bei einem nachfolgenden Besuchstermin nach dem ersten Anstieg des aINCAT-Wertes um ≥1 Punkt bestätigt wurde, oder ein Anstieg des aINCAT-Wertes um ≥ 2 Punkte im Vergleich zu Baseline von Phase B.
Die Studie wurde beendet, nachdem in der primären Endpunktanalyse 88 Ereignisse eines Rückfalls aufgetreten waren. Patient:innen aus dem Placebo-Arm konnten im Falle einer klinischen Verschlechterung in die offene Verlängerungsstudie ADHERE+# aufgenommen werden und VYVGART® erhalten. Die große Mehrheit der Betroffenen erklärte sich damit einverstanden. Ebenso hatten Patient:innen im Verum-Arm, die während der kontrollierten Phase stabil blieben, die Möglichkeit, im Rahmen von ADHERE+ weiterhin mit VYVGART® behandelt zu werden. 99 % aller in der ADHERE-Zulassungsstudie eingeschlossenen Patient:innen entschieden sich für die Fortführung von VYVGART® bzw. für eine Einleitung der Therapie mit VYVGART® in der ADHERE+ Studie.
Der primäre Endpunkt der Phase B ist die Zeit bis zur ersten klinischen relevanten Verschlechterung anhand des aINCAT-Scores. Der Anteil der Patient:innen, die einen Rückfall erlitten haben, war unter VYVGART® signifikant geringer als im Vergleich zu Placebo: Daraus ergibt sich eine hohe Wirksamkeit von VYVGART® bei der Vermeidung klinischer Rückfälle bei Patient:innen mit CIDP.
- Relative Risikoreduktion unter VYVGART® vs. Placebo: 61 %
- Rückfallrate: 27,9 % unter VYVGART® vs. 53,6 % unter Placebo
- Hazard Ratio (HR): 0,39 (95 % KI: 0,25–0,61; p = 0,0001)
VYVGART®: 61 % Reduktion des Rückfallrisikos vs. Placebo1,2
Primärer Endpunkt: Relatives Rückfallrisiko
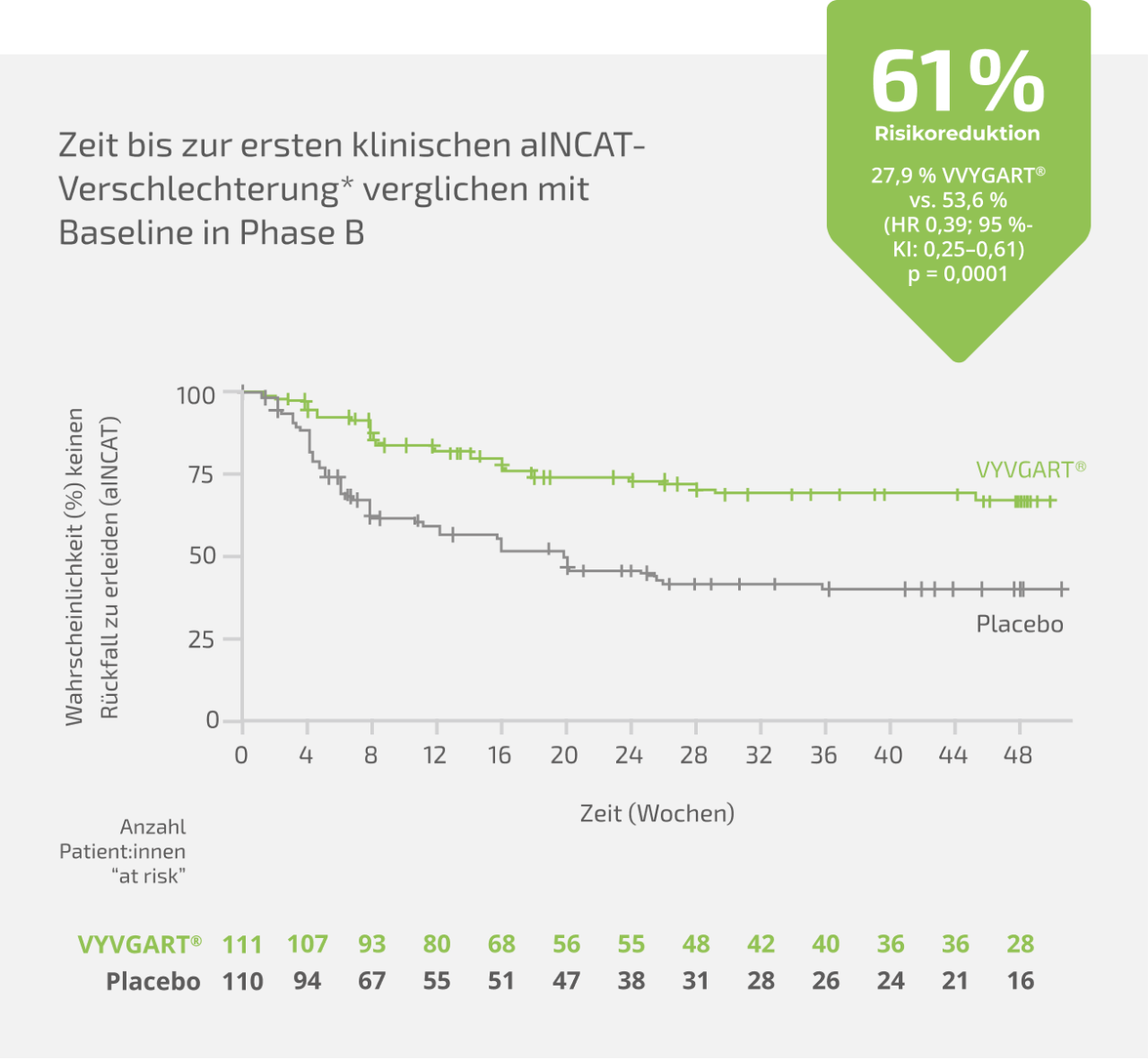
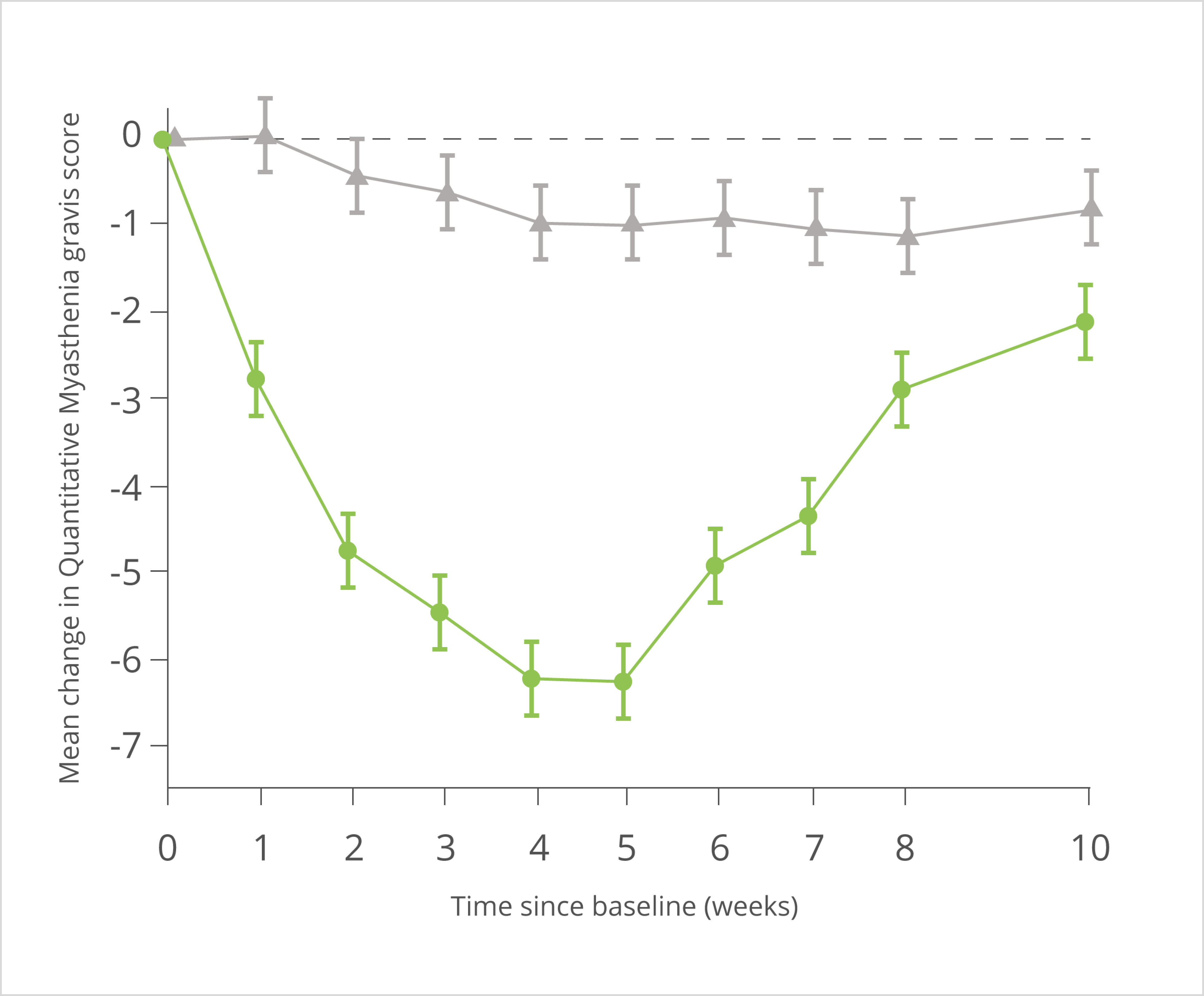
Ergebnisse der Gesamtpopulation
Die hier gezeigten Daten zeigen die gesamte ADHERE-Studienpopulation. VYVGART® wird als Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet.
* Primärer Endpunkt der randomisierten, doppelblinden, placebokontrollierten Phase B: Zeit bis zur ersten klinischen Verschlechterung (Rückfall), definiert als ein Anstieg des aINCAT-Wertes um 1 Punkt im Vergleich zu Baseline von Phase B, der bei einem nachfolgenden Besuchstermin nach dem ersten Anstieg des aINCAT-Wertes um 1 Punkt bestätigt wurde, oder ein Anstieg des aINCAT-Wertes um ≥ 2 Punkte im Vergleich zu Baseline von Phase B.
aINCAT: adjusted Inflammatory Neuropathy Cause and Treatment; HR: Hazard Ratio; KI: Konfidenzintervall
Auf der Basis dieser Ergebnisse wurde VYVGART® in Deutschland im Juni 2025 als Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Glukokortikosteroiden oder Immunglobulinen zugelassen.2
Wirkmechanismus von VYVGART® – Der erste innovative Wirkansatz seit 30 Jahren
DE-VYV-25-00116 / Mai 2026
VYVGART® ist ein rekombinantes, humanisiertes Fc-Fragment des humanen IgG1-Antikörpers, das zielgerichtet und passgenau mit einer erhöhten Affinität an die natürliche IgG-Bindungsstelle des neonatalen Fc-Rezeptors (FcRn) bindet.5,6 Der neonatale Fc-Rezeptor (FcRn) spielt eine zentrale Rolle im IgG-Metabolismus, indem er zirkulierendes IgG rezykliert und so dessen Halbwertszeit verlängert sowie die Serumkonzentration stabil hält.5,7 Unter physiologischen Bedingungen schützt FcRn IgG vor lysosomaler Degradation, indem es IgG in Endosomen bindet, rezirkuliert und so im Kreislauf hält.
Damit läutet VYVGART® eine neue Ära für die Behandlung von Patient:innen mit CIDP ein. Zum ersten Mal seit über 30 Jahren steht nun mit VYVGART® eine Therapie mit einem neuartigen Wirkmechanismus zur Behandlung der CIDP zur Verfügung.
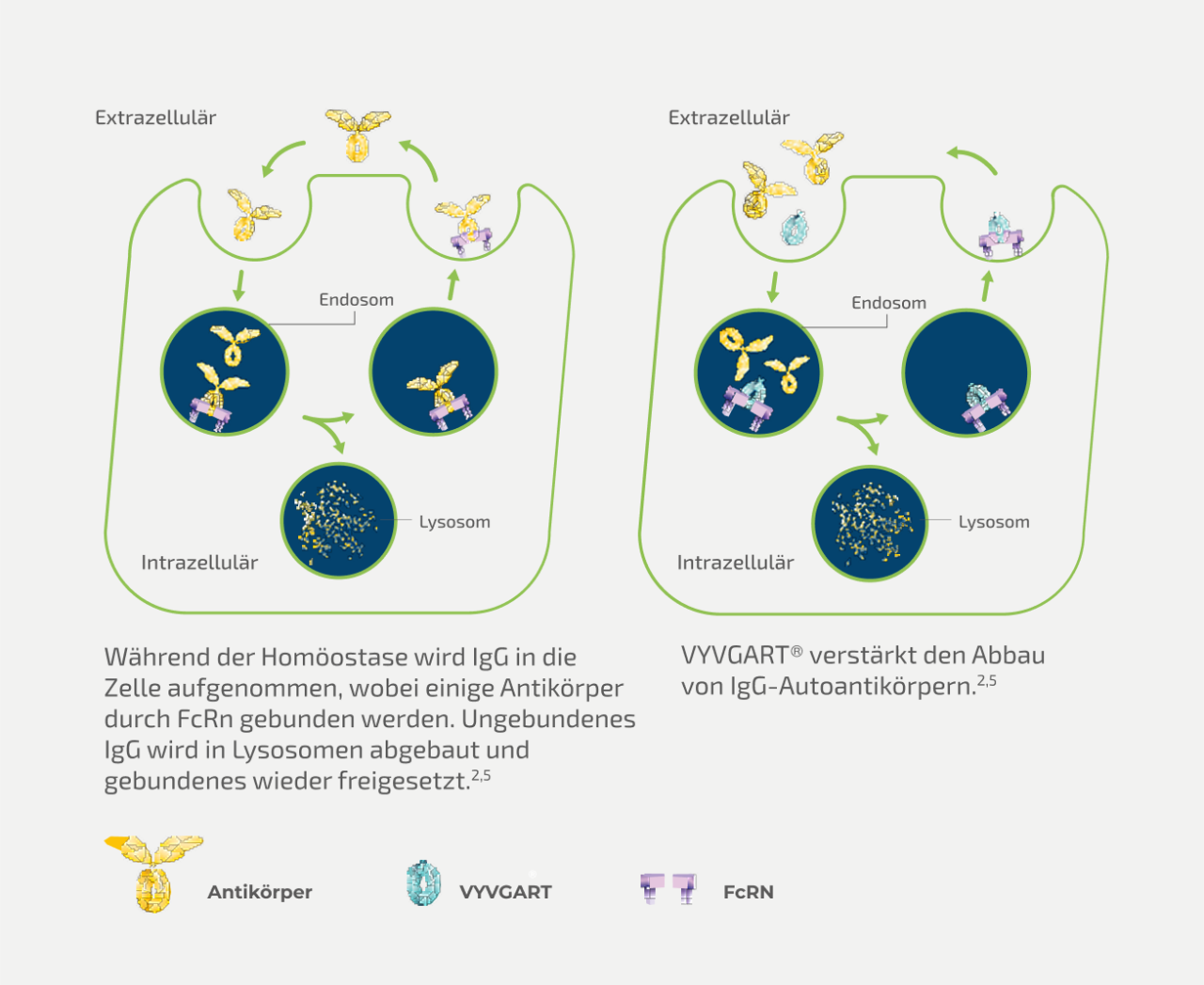
Die obere Abbildung zeigt den natürlichen Recyclingmechanismus von IgG-Antikörpern über den neonatalen Fc-Rezeptor (FcRn) (links) sowie die Wirkweise von VYVGART® (rechts). Normalerweise werden IgG-Antikörper über Endosomen in die Zelle aufgenommen. Dort bindet ein Teil an den FcRn und wird vor dem Abbau geschützt, während ungebundene IgGs im Lysosom abgebaut werden. VYVGART® blockiert gezielt die Bindung von IgG an FcRn. Dadurch wird weniger IgG recycelt und vermehrt im Lysosom abgebaut. Dies betrifft auch krankheitsverursachende IgG-Autoantikörper.
- VYVGART® wurde so konzipiert, dass es endogenes IgGs, und damit auch pathogene IgGs kompetitiv verdrängt, deren Recycling hemmt und stattdessen die lysosomale Degradation von IgGs fördert, ohne die IgG-Synthese direkt zu beeinflussen.5-7, 9, 10
- VYVGART® bindet ausschließlich an den neonatalen Fc-Rezeptor (FcRn): Als reines Fc-Fragment blockiert es nicht die Bindungsstellen für andere Immunglobuline wie IgM, IgA, IgE oder IgD und beeinflusst auch nicht die Albuminbindung am FcRn.
- Keine Beeinträchtigung von Albumin: Der Albuminspiegel bleibt stabil
- Keine Auswirkung auf Cholesterin: Auch der Lipidstoffwechsel wird nicht beeinflusst.
- Erhalt der Immunabwehr: Da andere Immunglobulin-Subklassen, wie IgN, IgA, IgE oder IgD nicht beeinträchtigt werden, erhöht sich auch nicht das Risiko für Infektionen. Eine routinemäßige Impfanpassung ist nicht erforderlich.2
VYVGART® wirkt damit genau dort, wo es therapeutisch notwendig ist. Und das mit einem guten Sicherheitsprofil, weil es die körpereigene Abwehr nicht pauschal unterdrückt, sondern selektiv auf IgG Moleküle und damit auch auf krankheitsrelevante IgG-Autoantikörper abzielt.
VYVGART® in Kombination mit dem Enzym Hyaluronidase (PH20) ermöglicht eine effiziente subkutane Applikation, da PH20 die Resorption aus dem subkutanen Gewebe beschleunigt. Nach Aufnahme in den systemischen Kreislauf bindet Efgartigimod alfa an den neonatalen Fc-Rezeptor (FcRn) auf Endothelzellen und blockiert so das Recycling von IgG.5,6,11,13
- Das subkutane Gewebe enthält natürlicherweise Hyaluronan, eine Substanz, die das Gewebe strukturiert, aber auch die Durchlässigkeit für Medikamente begrenzt. Genau hier setzt PH20 an.
- Die Hyaluronidase spaltet lokal das Hyaluronan auf und macht das Gewebe kurzfristig durchlässiger.13
- Dadurch kann VYVGART® leichter vom Gewebe ins Blutgefäßsystem gelangen.
- Die normale Gewebestruktur bildet sich innerhalb von 24 bis 48 Stunden wieder vollständig zurück.13,14 Der Effekt ist also reversibel und gut kontrolliert.
- Einmal im Blutkreislauf angekommen, bindet VYVGART® gezielt an den neonatalen Fc-Rezeptor (FcRn) auf den Endothelzellen. Das verhindert den natürlichen Recyclingprozess von Immunglobulin G (IgG), einschließlich krankheitsverursachender Autoantikörper.
- Diese werden dadurch vermehrt abgebaut, ohne andere Immunglobuline wie IgA, IgM oder IgE zu beeinflussen.
* VYVGART® wird als Monotherapie zur Behandlung von erwachsenen Patient:innen mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet.2
+ Definition aktive CIDP in der ADHERE-Studie: Um sicherzustellen, dass nur Patient:innen mit aktiver Erkrankung in die ADHERE-Studie eingeschlossen wurden, musste in der max. 12-wöchentlichen Run-in-Phase, in der die Vormedikation abgesetzt wurde, eine klinisch relevante Verschlechterung (Evidence of Clinical Meaningful Deterioration, ECMD) nachgewiesen werden. Dazu musste mindesten eines der folgenden Kriterien erfüllt sein:
- Anstieg des aINCAT-Scores um ≥ 1 Punkt
- Abfall des I-RODS um ≥ 4 Punkte
- Reduktion der Handkraft um ≥ 8 kPa (Martin-Vigorimeter)
# Aufnahmeberechtigt in ADHERE+ waren alle Teilnehmenden, die entweder nach 48 Wochen Phase B stabil geblieben waren, einen Rückfall erlitten oder zum Zeitpunkt des Erreichens des Primärendpunkts in der offenen Phase oder Run-in-Phase waren. Fast 99 % der berechtigten Patient:innen (n=228 von 229) entschieden sich, VYVGART® in ADHERE+ weiterhin zu erhalten.
1 Howard JF, Jr., Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-36.
2 Fachinformation VYVGART® (Stand Juni 2025)
3 Allen JA, Lin J, Basta I, Dysgaard T, Eggers C, Guptill JT, et al. Safety, tolerability, and efficacy of subcutaneous efgartigimod in patients with chronic inflammatory demyelinating polyradiculoneuropathy (ADHERE): a multicentre, randomised-withdrawal, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2024;23(10):1013-24.
4 Van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Vankrunkelsven P, Allen JA, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision. Eur J Neurol. 2021;28(11):3556-83.
5 Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372-86.
6 Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23(10):1283-8.
7 Sesarman A, Vidarsson G, Sitaru C. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol Life Sci. 2010;67(15):2533-50.
8 Howard JF, Jr., Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-36.
9 Nixon AE, Chen J, Sexton DJ, Muruganandam A, Bitonti AJ, Dumont J, et al. Fully human monoclonal antibody inhibitors of the neonatal fc receptor reduce circulating IgG in non-human primates. Front Immunol. 2015;6:176.
10 Ward ES, Gelinas D, Dreesen E, Van Santbergen J, Andersen JT, Silvestri NJ, et al. Clinical Significance of Serum Albumin and Implications of FcRn Inhibitor Treatment in IgG-Mediated Autoimmune Disorders. Front Immunol. 2022;13:892534.
11 Wolfe GI, Ward ES, de Haard H, Ulrichts P, Mozaffar T, Pasnoor M, et al. IgG regulation through FcRn blocking: A novel mechanism for the treatment of myasthenia gravis. J Neurol Sci. 2021;430:118074.
12 Mathey EK, Park SB, Hughes RA, Pollard JD, Armati PJ, Barnett MH, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86(9):973-85.
13 Locke KW, Maneval DC, LaBarre MJ. ENHANZE(®) drug delivery technology: a novel approach to subcutaneous administration using recombinant human hyaluronidase PH20. Drug Deliv. 2019;26(1):98-106.
14 Wasserman RL, Melamed I, Stein MR, Engl W, Sharkhawy M, Leibl H, et al. Long-Term Tolerability, Safety, and Efficacy of Recombinant Human Hyaluronidase-Facilitated Subcutaneous Infusion of Human Immunoglobulin for Primary Immunodeficiency. J Clin Immunol. 2016;36(6):571-82.
Pflichtangaben DE nach § 74a AMG
Vyvgart 20 mg/ml Konzentrat zur Herstellung einer Infusionslösung/
Vyvgart 1 000 mg Injektionslösung in einer Fertigspritze
▼ Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Bezeichnung: Vyvgart 20 mg/ml Konzentrat zur Herstellung einer Infusionslösung/Vyvgart 1 000 mg Injektionslösung in einer Fertigspritze. Wirkstoff: Efgartigimod alfa. Pharmakotherapeutische Gruppe: Immunsuppressiva, selektive Immunsuppressiva, ATC-Code: L04AA58. Qualitative und quantitative Zusammensetzung: Vyvgart 20 mg/ml: Jede Durchstechflasche mit 20 ml enthält 400 mg Efgartigimod alfa (20 mg/ml). Sonstige Bestandteile: Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Natriumchlorid, Saccharose, Argininhydrochlorid, Polysorbat 80 (E433), Wasser für Injektionszwecke. Jede Durchstechflasche enthält 33,2 mg Natrium. Jede Durchstechflasche enthält 8,2 mg Polysorbat 80, entsprechend 0,4 mg/ml. / Vyvgart 1 000 mg Injektionslösung in einer Fertigspritze: Jede Fertigspritze enthält 1 000 mg Efgartigimod alfa in 5 ml (200 mg/ml). Sonstige Bestandteile: Vorhyaluronidase alfa, Argininhydrochlorid, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80 (E433), Natriumchlorid, Saccharose, Wasser für Injektionszwecke. Jede Fertigspritze enthält weniger als 1 mmol (23 mg) Natrium. Jede Fertigspritze enthält 2,1 mg Polysorbat 80, entsprechend 0,4 mg/ml. Anwendungsgebiete: Vyvgart 20 mg/ml: Vyvgart wird zusätzlich zur Standardtherapie zur Behandlung von erwachsenen Patienten mit generalisierter Myasthenia gravis (gMG) angewendet, die Anti-Acetylcholin-Rezeptor (AChR)-Antikörper positiv sind. / Vyvgart 1 000 mg Injektionslösung in einer Fertigspritze: Vyvgart wird zusätzlich zur Standardtherapie zur Behandlung von erwachsenen Patienten mit generalisierter Myasthenia gravis (gMG) angewendet, die Anti-Acetylcholin-Rezeptor (AChR)-Antikörper positiv sind. Vyvgart wird als Monotherapie zur Behandlung von erwachsenen Patienten mit progredienter oder rezidivierender aktiver chronisch-entzündlicher demyelinisierender Polyneuropathie (CIDP) nach vorheriger Behandlung mit Kortikosteroiden oder Immunglobulinen angewendet. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der oben genannten sonstigen Bestandteile. Nebenwirkungen: Vyvgart 20 mg/ml: Sehr häufig: (≥ 1/10): Infektionen der oberen Atemwege; Häufig (≥ 1/100, < 1/10): Harnwegsinfektionen, Bronchitis, Myalgie, Kopfschmerz im Zusammenhang mit dem Verfahren, Übelkeit; Nicht bekannt: anaphylaktische Reaktion während oder nach der Infusion. / Vyvgart 1 000 mg Injektionslösung in einer Fertigspritze: Sehr häufig: (≥ 1/10): Infektionen der oberen Atemwege, Reaktionen an der Injektionsstelle; Häufig (≥ 1/100, < 1/10): Harnwegsinfektionen, Bronchitis, Myalgie, Übelkeit. Warnhinweise: Arzneimittel für Kinder unzugänglich aufbewahren. Verschreibungspflichtig. Pharmazeutischer Unternehmer/ Inhaber der Zulassung: argenx BV, Industriepark-Zwijnaarde 7, 9052 Gent, Belgien.
Stand: Januar 2026.
DE-VYV-25-00128
X
Hinweis: Aus rechtlichen Gründen dürfen Informationen zu verschreibungspflichtigen Medikamenten nur medizinischen Fachkreisen oder Patient*innen mit einer Verschreibung für dieses Medikament zugänglich gemacht werden. Bei Fragen zu Therapien wenden Sie sich bitte an Ihre Ärztin bzw. Ihren Arzt.
X
X
Da Sie keine Verschreibung für dieses Medikament haben und auch nicht den medizinischen Fachkreisen angehören, sind wir aus rechtlichen Gründen nicht befugt, Ihnen Informationen bereitzustellen. Sollten Sie Fragen zu Therapien haben, empfehlen wir Ihnen, sich an Ihre Ärztin bzw. Ihren Arzt zu wenden.
Besuchen Sie gerne unsere Websites für Patient:innen und Angehörige:
X